Importing MSFragger results (including timsTOF PASEF) into Skyline
This tutorial covers spectral library building in Skyline from validated MSFragger search results. In this example, we used Skyline-daily (v 19.1.9.350) and two files from the timsTOF PASEF dataset published by Meier et al, which can be downloaded from ProteomeXchange (‘HeLa_200ng_100ms_raw.zip’ on the FTP site).
This tutorial can also be used to import Thermo data into Skyline, ‘.mzML’ files can be used in place of ‘.d’ and ‘.mgf’/’_calibrated.mgf’. Additionally, results from DIA (‘interact-pep.xml’ files and pseudo-DDA ‘.mzML’ files generated by the ‘DIA-Umpire’ workflow in FragPipe can also be imported into Skyline using this method.
Set up the analysis
Make sure the PeptideProphet output files (‘interact-pep.xml’), raw spectral files (‘.d’, ‘.mzML’, or ‘.raw’), and .mgf files (‘_uncalibrated.mgf’) are in the same directory or in the same parent directory. You will also need the protein sequence database filtered to 1% FDR (‘protein.fas’) written by Philosopher.
Create a new DDA document
Launch Skyline and select ‘Import DDA Peptide Search’ from the Start Page. Follow the prompts to choose a name and location for the new document to be saved.
Import peptide results
Once the new document is saved, ‘Add Files’ to select the ‘interact-pep.xml’ files for the search results you want to import.
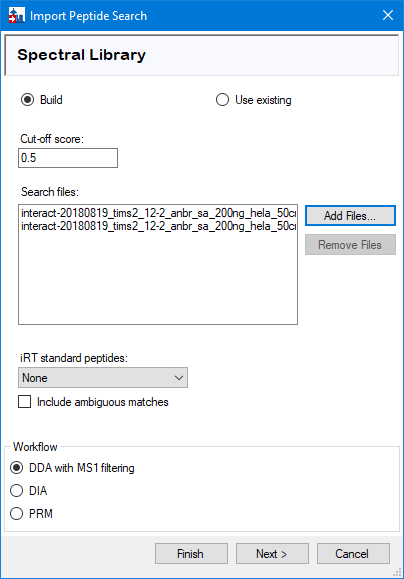
To generate a spectral library that is filtered to 1% peptide FDR and 1% protein FDR, we recommend the following:
1) Specifying the peptide probability threshold that corresponds to 1% peptide FDR as the ‘Cut-off score’. (This can be found in the FragPipe log or .log file, a cut-off of 0.9024 would be used in this example: INFO[15:56:25] Converged to 1.00 % FDR with 57143 Peptides decoy=576 threshold=0.9024 total=57719)
2) Using ‘protein.fas’, which is generated by Philosopher filtering and contains sequences of proteins that passed 1% protein-level FDR file, in the ‘Add FASTA file’ step below.
Once you’ve set a cut-off score, press ‘Next’, prompting Skyline to build the spectral library from these peptide search results.
Add spectral files
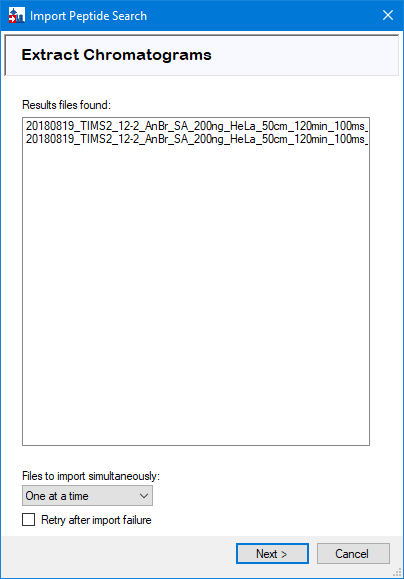
When the spectral library is built, Skyline should automatically detect the spectral files. Press ‘Next’. If Skyline detects that the files in your experiment are named with a common prefix, you have the option to shorten the result names at this point for easier comparison later on.
Add detected modifications
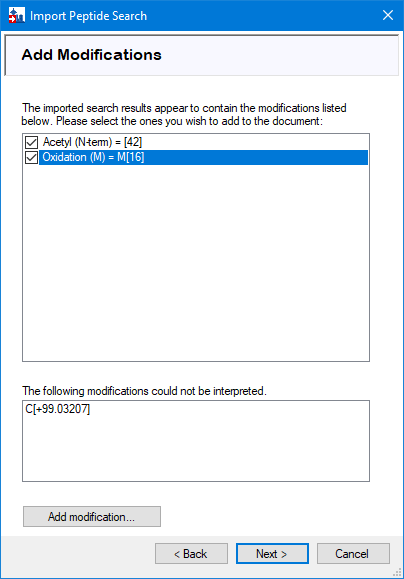
Skyline should automatically detect variable modifications from your search results. Select the suggested modifications and press ‘Next’.
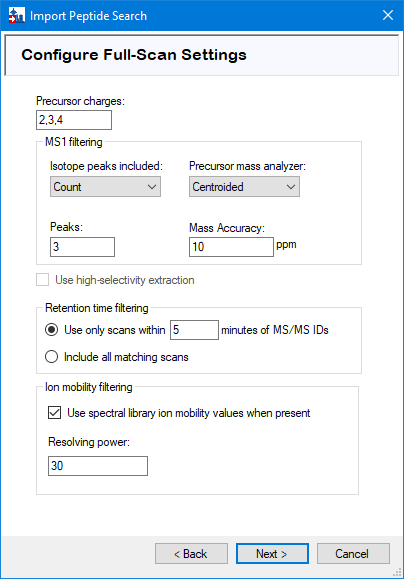
Configure scan settings
Precursor charge states greater than 2 can be added at this point. Press ‘Next’.
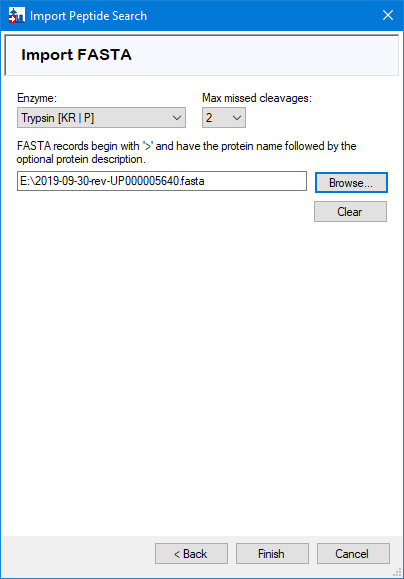
Add FASTA file
‘Browse’ for the sequence database, and select the number of missed cleavages that was used in the database search. Press ‘Next’ to insert the ‘protein.fas’ file output by FragPipe/Philosopher, which contains the sequences of proteins filtered to 1% FDR.
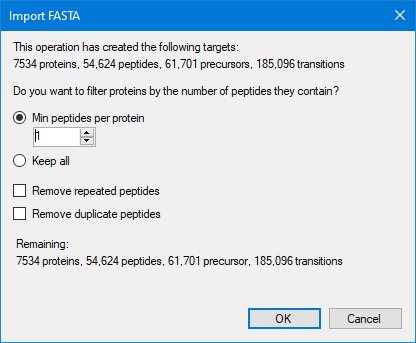
Filter targets
When the FASTA file is finished importing, select the desired filters, and press ‘OK’ to start the import.
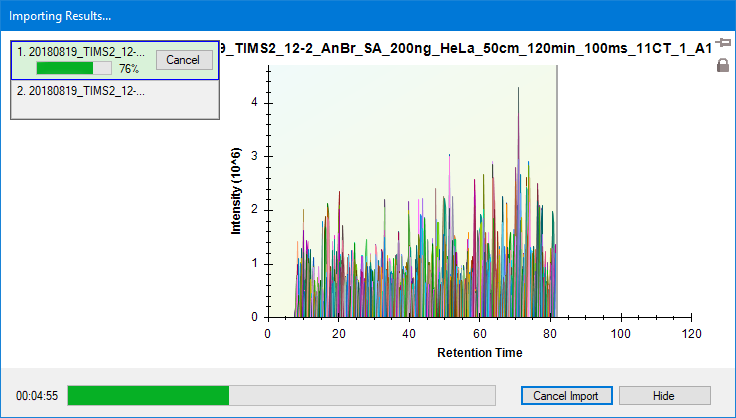
Importing the results
The import will take a few minutes per file.
View results
Once the import is finished, you are ready to perform your Skyline analyses, select a peptide from the list to view. Quantification can be compared between runs via View > Peak Areas > Replicate Comparison.
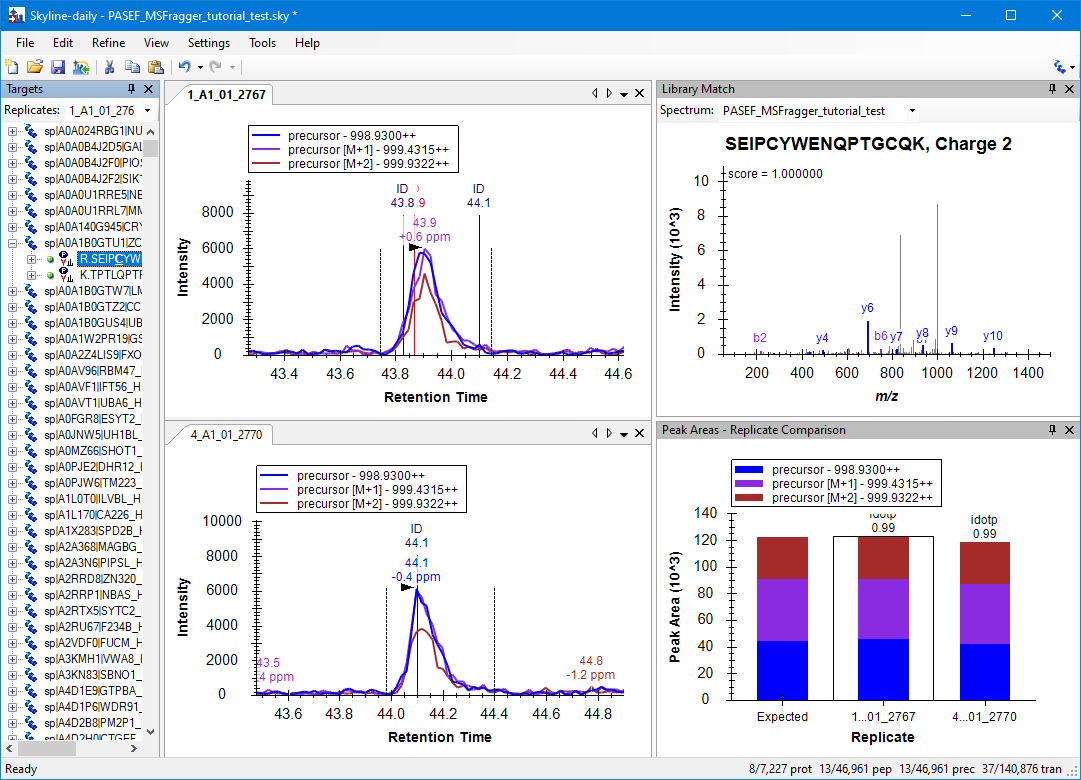
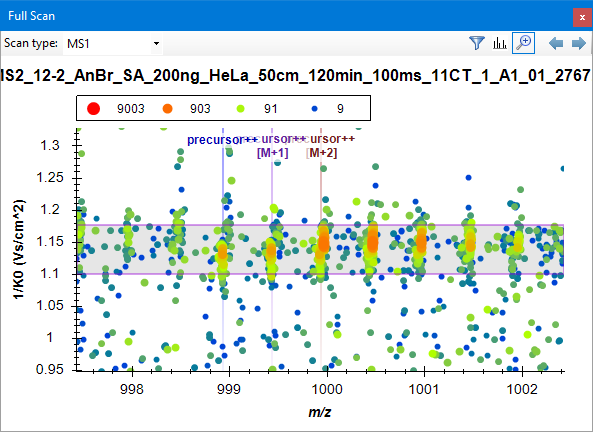
To see the two-dimensional ion mobility vs. m/z plot, click on the apex of the chromatogram.