Using a custom offset workflow with FragPipe: RNA crosslinking analysis
XRNAX is a method for “unbiased” purification of protein-crosslinked RNA, which can be used to help probe and understand protein-RNA interactions. For this analysis, we will need to load a custom workflow. This workflow searches the database with mass offsets that correspond to crosslinked RNA fragments. One can alternatively perform a fully open search (with the mass window set to -150, +1000 Da) as was done in the publication (citation below). However, mass offset searches are faster and more sensitive than performing a full open search, with the limitation that you can only find peptides with mass shifts specified in the offset list.
This tutorial will use ‘XRNAX-MassOffset’ workflow to search a single .mzML spectral file of RNA-crosslinked tryptic peptides from an MCF7 cell line, acquired on a QExactive HF. Original publication: Trendel, Jakob, et al. “The human RNA-binding proteome and its dynamics during translational arrest.” Cell 176.1-2 (2019): 391-403. The 20160530_QE1_JT_XRNAXpep_Trp_hydro.mzML you will need for this tutorial can be found from PRIDE.
Tutorial contents
- Load a custom workflow
- Inspect the search parameters
- Get a sequence database
- Set the custom annotation file
- Pick an output location and run
- Examine the results
Load a custom workflow
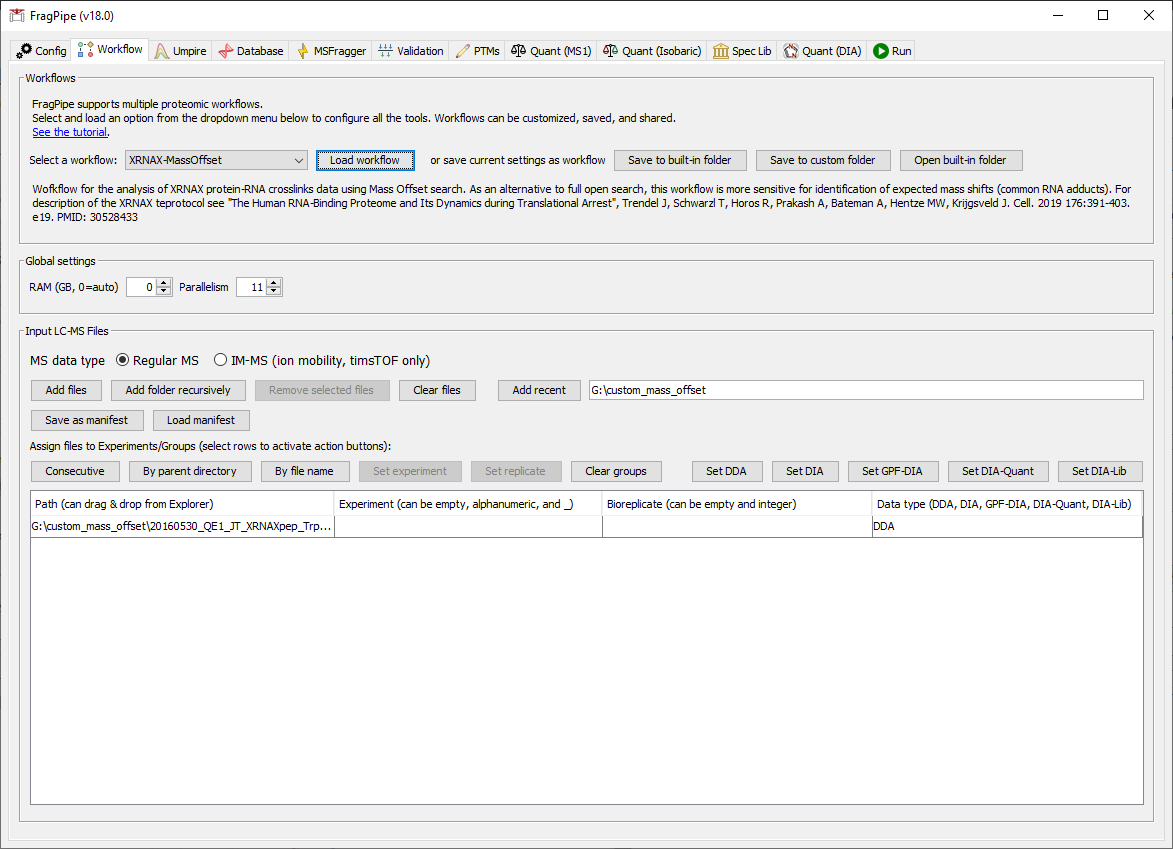
On the Workflow tab, select ‘XRNAX-MassOffset’ from the workflow dropdown menu and press ‘Load’. Drag and drop or use ‘Add files’ to input the file 20160530_QE1_JT_XRNAXpep_Trp_hydro.mzML.
Get a sequence database
You can download a human protein database file using FragPipe. On the Database tab, click the ‘Download’ button. Follow the prompts to use the default settings (reviewed human sequences with common contaminants).
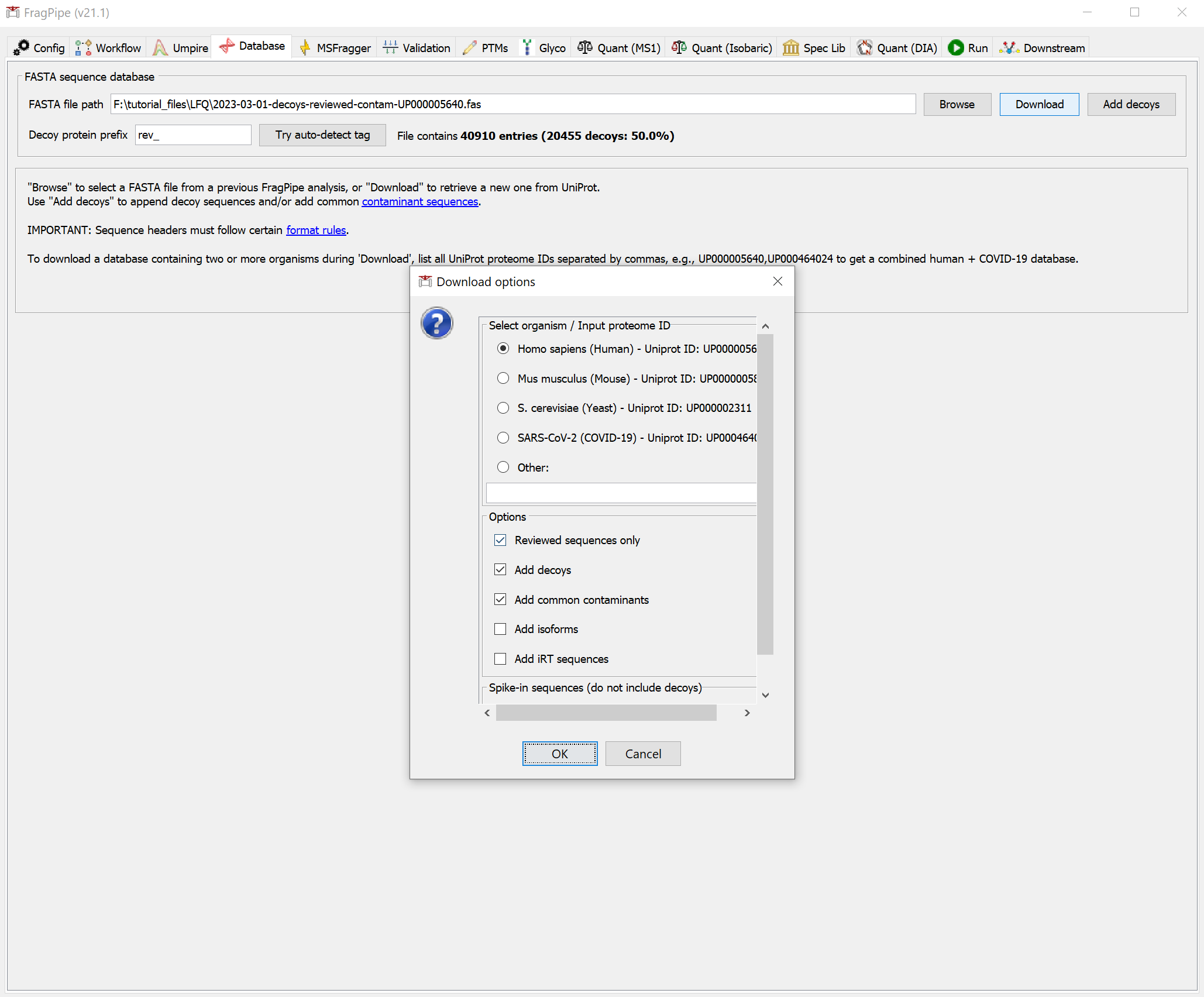
Click ‘Yes’ to download the database. When it’s finished, you should see that the FASTA file path now points to the new database.
Inspect the search parameters
On the MSFragger tab, inspect the search parameters. Scroll down to the Mass Offsets box in the Advanced Options section– this is where the crosslinked RNA fragment masses are specified.
Set the custom annotation file
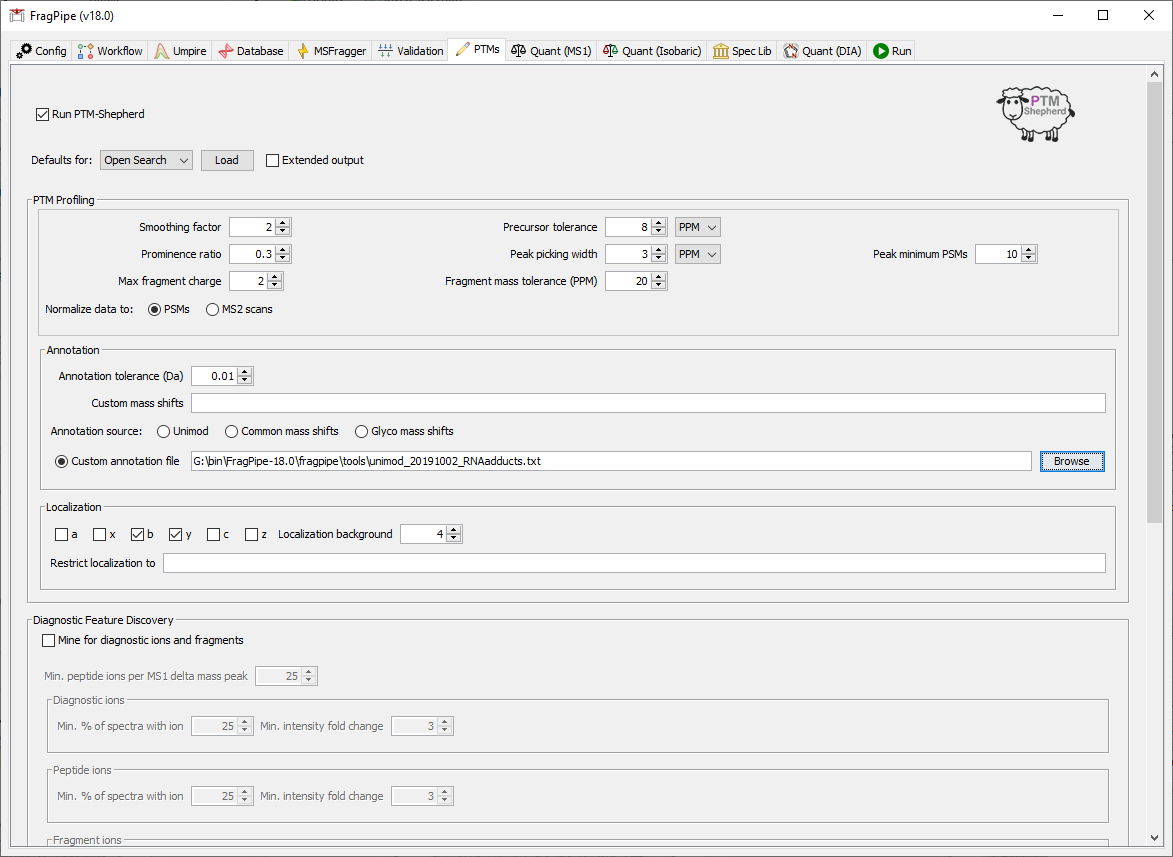
On the PTMs tab, we will add a custom annotation file to supply names for the mass offset values (as a list of name-mass pairs), download ‘unimod_20191002_RNAadducts.txt’ from here if you haven’t already. In the ‘Custom annotation file’ box, provide the file path to unimod_20191002_RNAadducts.txt. You can copy and paste the file path or ‘Browse’ to add it.
Pick an output location and run
Now we can start the analysis from the ‘Run’ tab after specifying an output location, you can make a new folder: ‘fragpipe_RNA-crosslinking_results’.
Examine the results
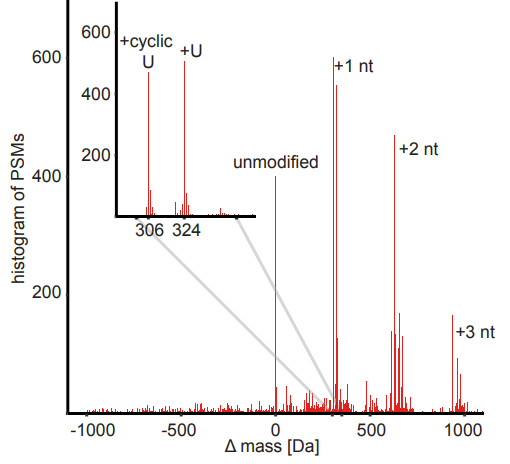
Inspect the global.profile.tsv file generated by PTM-Shepherd and compare it to the figure below (generated from the pepxml file with DeltaMass). This is a histogram of the full range of delta masses observed from all peptide-spectrum matches. Two of the most prominent peaks are labeled (U and cyclic-U), and the higher mass peaks (between 600-700 Da and 900-1015 Da) correspond to various combinations of two and three attached nucleotides, respectively.
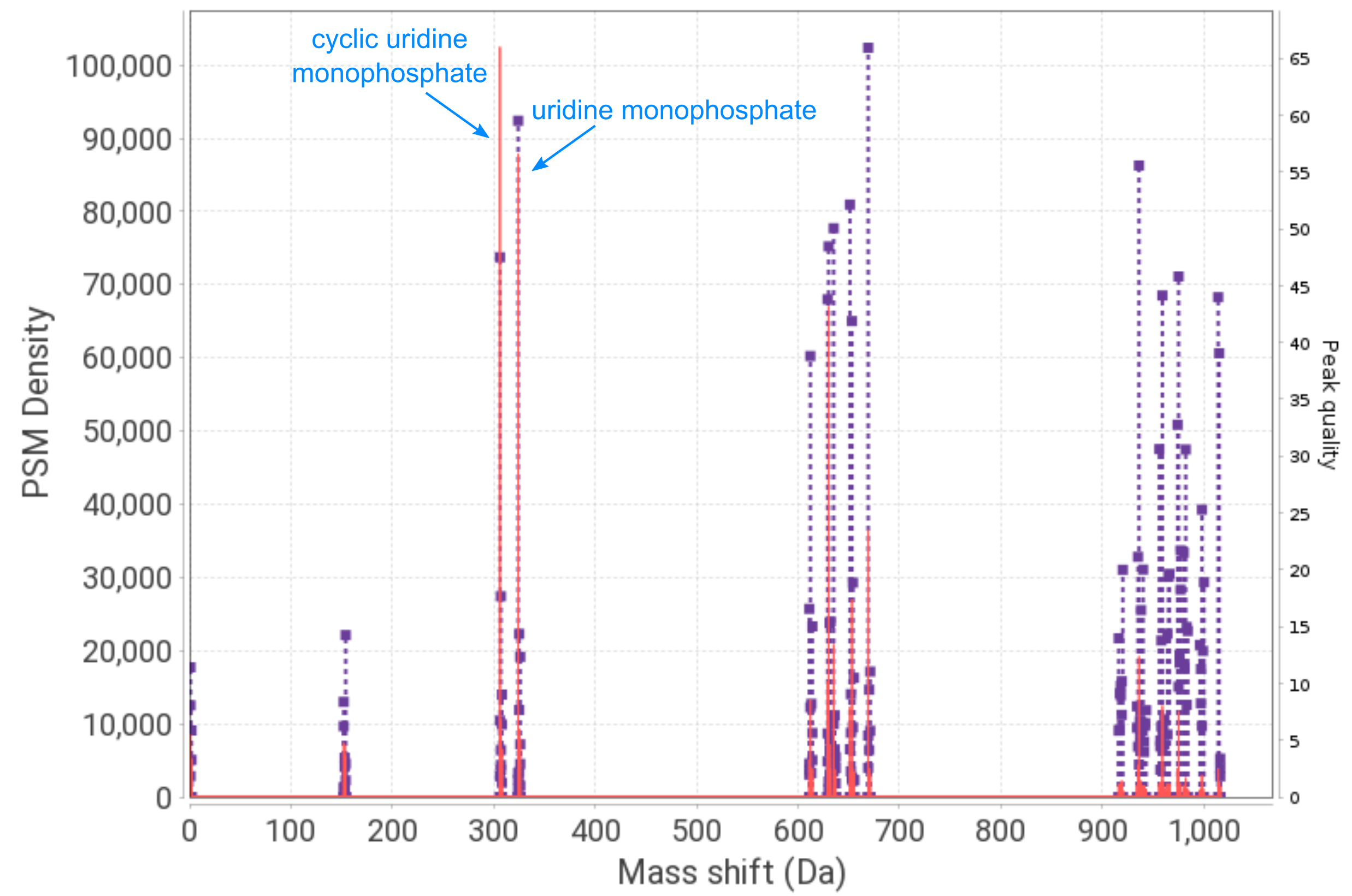
In the global.profile.tsv file, you should observe the mass shifts discussed in the manuscript (Figure 2B, shown below). Uridine phosphodiester (cyclic U) and uridine monophosphate (U, ‘Simulate peptide-RNA conjugates’ in Unimod) are the top two entries in the table. In the AA1 and AA1_enrichment_score columns, we can see that the uridine phosphodiester mass shift predominantly localizes to phenylalanine, consistent with the enrichment observed in the manuscript. Looking further down in the table, we can see that uridine phosphodiester also shows up in the mapped_mass_2 column alongside the first and second isotopic peaks.