Contents
- Offset search for PTMs with FragPipe
- Types of Mass Offset Searches
- Options for Reporting Mass Offsets
- Example Mass Offset Search Tutorial (tubulin search)
- Histone Detailed Mass Offset (DMO) Search using the HiP-Frag method
Offset search for PTMs with FragPipe
Traditional closed search requires that precursor masses match the identified peptide within a small mass tolerance (e.g., +/-20 ppm), often with variable modifications allowed on only one or a few amino acid residues. In contrast, the open search strategy allows precursor masses to differ from the identified peptide by any mass within a large range (e.g., -100 to +150). Mass offset search is an intermediate strategy, allowing selected mass differences (within a tolerance, e.g. 20 ppm) on any peptide match (far right, below).

The key difference between mass offset (and open) searches vs closed search is that only one mass offset is allowed per peptide, whereas multiple variable modifications can be searched on a peptide. This prevents combinatorial expansion of the search space and allows searching for many masses (e.g., PTMs) at once.
Types of Mass Offset Searches
Mass offset searches in MSFragger can be done without restriction, allowing offsets on any amino acid in any peptide, or with offsets restricted to specific amino acid(s). Labile mode diagnostic and remainder ions can also be specified (see the labile mode tutorial for more details).
There are 3 types of mass offset searches:
- Unrestricted: the traditional mass offset search
- Restricted: all offsets share the same restrictions (introducted in MSFragger v3.8)
- Restricted-detailed: each offset has its own set of restrictions (introduced in MSFragger v4.0)
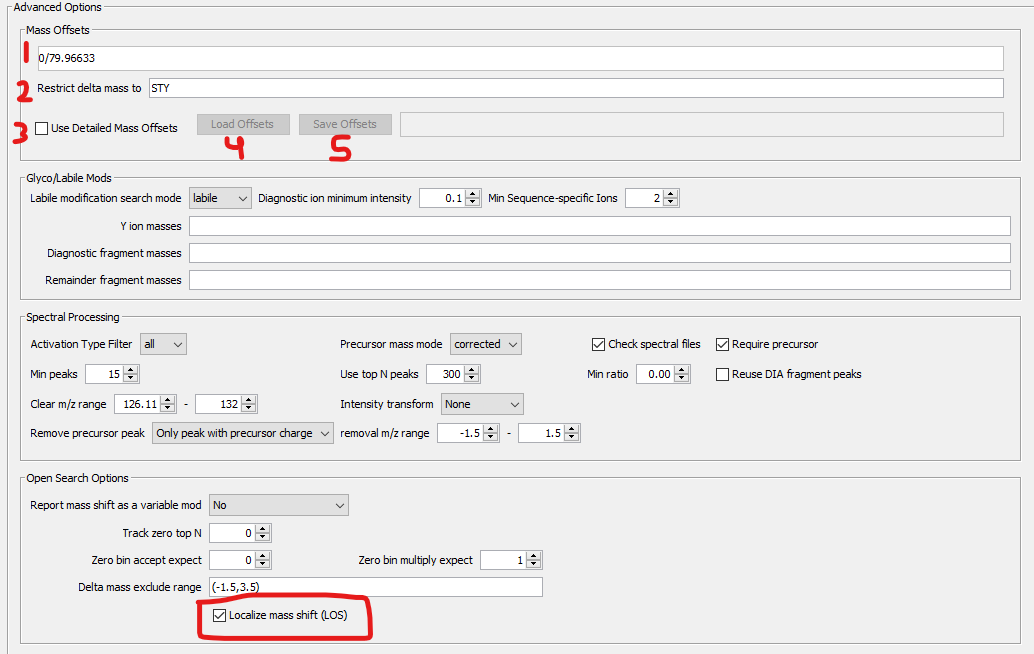
Unrestricted: To perform a regular unrestricted offset search, enter the mass offset masses of interest in the mass offsets box on the MSFragger tab (box 1, below).
Restricted: To perform a restricted offset search, enter the amino acid site(s) at which to allow mass offsets in the “Restrict delta mass to” box on the MSFragger tab (box 2, below). Note that ALL OFFSETS will have the same site restriction if multiple are entered. Labile mode ions can also be specified for offsets in the Glyco/labile Mods box below. NOTE: “Localize mass shift (LOS)” in the Open Search Options box must be checked for offsets to be localized to specific amino acids.
Restricted-detailed: To perform a detailed restricted offset search, check the “Use Detailed Mass Offsets” box (box 3, below). This will disable the regular mass offsets boxes 1 and 2, and enable the Load Offsets and Save Offsets buttons (4 and 5, below). Offsets can be loaded from a template file, which is a tab separated text file containing the offset mass, allowed amino acid(s), labile diagnostic ions, labile peptide remainder masses, and labile fragment remainder masses. Any field except mass can be left empty to ignore it. An example template file is shown below, and can be downloaded and used as a starting point.
Example detailed offset template file screenshot and link. Use “save link as” to download the example template file from the link below the image.
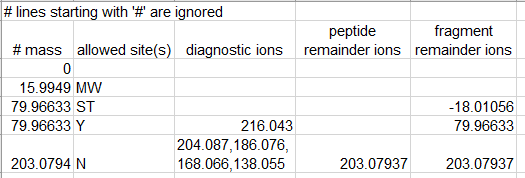
Example template file
Options for Reporting Mass Offsets
The Report mass shift as a variable mod option in the Open Search Options box can be used to determine where mass offsets are saved to the result files. This can enable, for example, quantification and site reporting of mass offsets as is done with variable modifications. However, there are implications for how the offsets are processed by the validation tools so choosing the correct setting is important. There are 3 settings available:
- No
- Yes, remove delta mass
- Yes, keep delta mass
No: The original method. Mass offsets are reported in the “Delta Mass” column of the psm.tsv table. PeptideProphet extended mass model CAN be used. PTM-Shepherd CAN process the results. Quantification of offsets is NOT possible.
Yes, remove delta mass: Mass offsets are subtracted from the Delta Mass column and added to the Assigned Modifications column of the psm.tsv (alongside any variable modifications found). PeptideProphet extended mass model CANNOT be used. PTM-Shepherd CANNOT process the results. Quantification of offsets IS possible.
Yes, keep delta mass: Mass offsets are added to the Assigned Modifications column of the psm.tsv, but are NOT subtracted from the Delta Mass column (and are thus reported twice). PeptideProphet extended mass model CAN be used, and PTM-Shepherd CAN process the results. Label-free quantification of the offsets is NOT possible unless there is additional downstream processing (e.g., as done in glyco searches) that removes the delta mass prior to IonQuant. Isobaric labeled quantification IS possible using TMT-Integrator.
For discovery searches (e.g., looking for many modifications as mass offsets, like the MassOffsets-CommonPTMs workflow), No is typically the preferred option as quantification is not required and extended mass modeling in PeptideProphet and processing by PTM-Shepherd can be important. For searches considering a small number of offsets or if quantification is desired, one of the Yes options is typically preferred: keep delta mass if extended mass modeling in PeptideProphet is required, or remove delta mass if not. Extended mass modeling is typically important if there is a wide range of abundances of offsets (e.g., in glyco searches where some glycans are extremely rare while others are quite common).
Example Mass Offset Search Tutorial
An example walking through a traditional mass offset search is shown here. Parthenolide is a protein-reactive compound frequently used by cytoskeleton researchers to inhibit tubulin detyrosinases, but it is suspected to hit more than just those enzymes. Labeled peptides should have a +248.14125 Da mass shift, and we want to allow modification of any amino acid so we can learn more about parthenolide’s specificity. This is a case for mass offset search.
The samples we will use for this tutorial are purified tubulin treated with either DMSO (control) or parthenolide, download PRF_Q_2019_R_OHI_51_47069.mzML and PRF_Q_2019_R_OHI_51_47070.mzML from PRIDE. Citation: Hotta, Takashi, et al. “Parthenolide Destabilizes Microtubules by Covalently Modifying Tubulin.” Current Biology 31.4 (2021): 900-907.
Example contents
- Add the data
- Fetch a sequence database
- Customize the search settings
- Set the output location and run
- Examine the results
Open FragPipe
When you launch FragPipe, check that MSFragger, IonQuant, and Philosopher are all configured. If you haven’t downloaded them yet, use their respective ‘Download / Update’ buttons. Please see the tutorials here and here for more help. Python is not needed for this exercise.
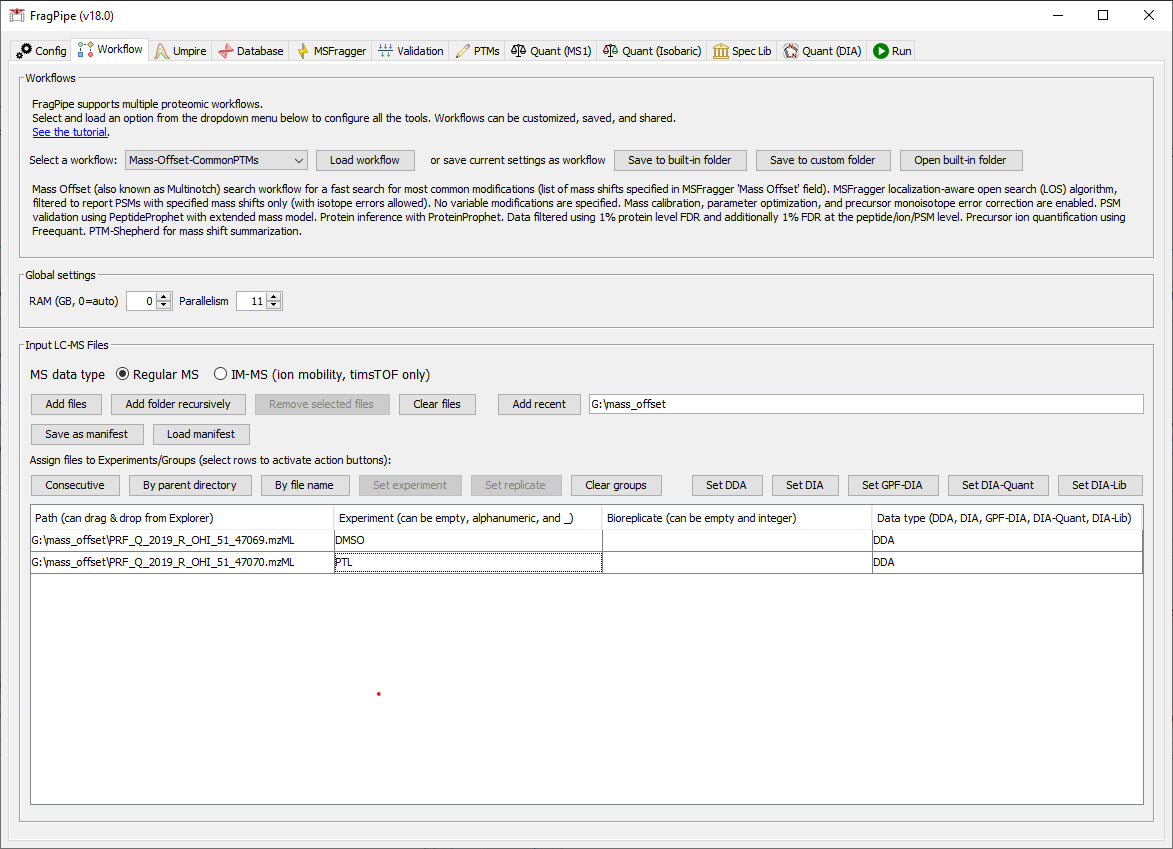
Add the data
1) Load the mzML files and specify experiments: “DMSO” for the file ending 69, “PTL” (short for parthenolide) for the one ending 70. 2) Select the ‘Mass-Offset-CommonPTMs’ workflow from the dropdown menu and click ‘Load’.
Fetch a sequence database
3) Now we need to select a protein sequence database. You can download a human .fas file using FragPipe. Downloading is easy, so we could also choose to download one at this point. On the Database tab, click the ‘Download’ button. Follow the prompts to use the default settings (reviewed human sequences with common contaminants).
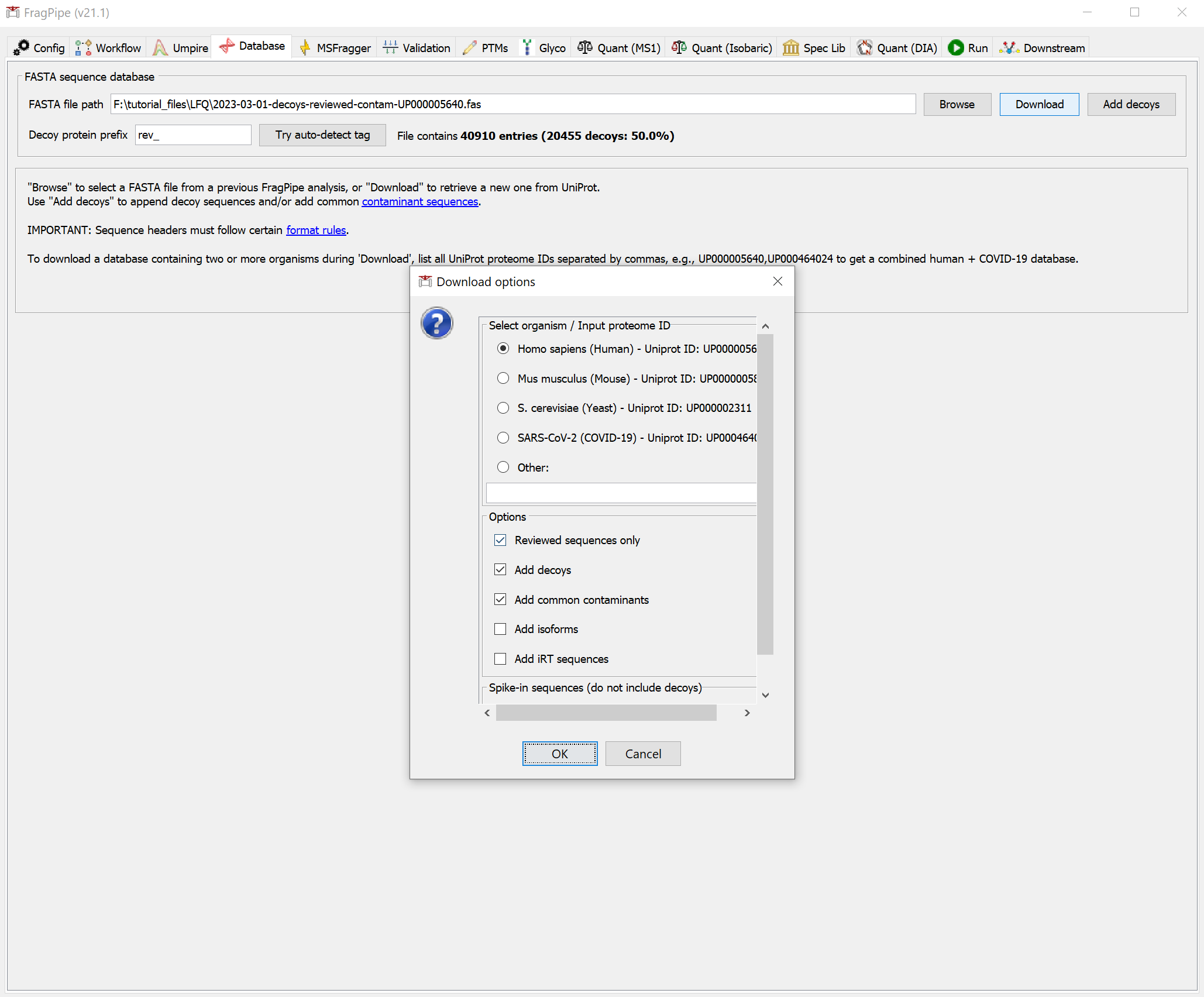
Click ‘Yes’ to download the database. When it’s finished, you should see that the FASTA file path now points to the new database.
Customize the search settings
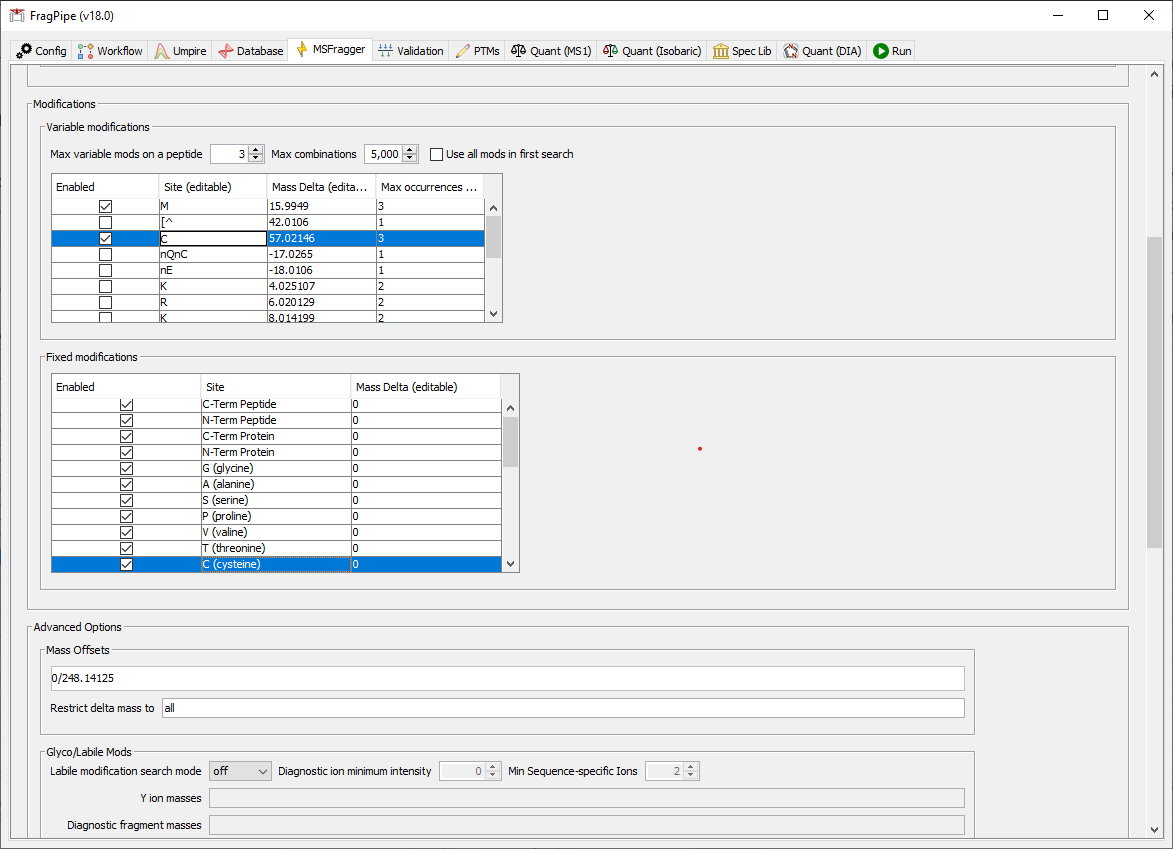
4) In the MSFragger tab, check the first box in the Variable modifications section to set methionine oxidation as a variable modification. Because we want to allow parthenolide to label any residue (and because cysteines were alkylated after parthenolide was added), we need to make cysteine carbamidomethylation a variable mod instead of a fixed mod. To do this, first add C +57.02146 to the list of variable modifications.
5) Then scroll down to set fixed Mass Delta to 0 for cysteine.
6) When we loaded the ‘Mass-Offset-CommonPTMs’ workflow, the default list of offsets includes more than we need for this analysis since we want to focus on parthenolide. In the ‘Mass Offsets’ field, replace the offsets list with just 0/248.14125 for the parthenolide adduct. The list of mass offsets can be / or space separated, and only the specified mass offsets will be allowed in the search (i.e. combinations of specified mass offsets will not be generated).
Please note: Only peptides with the mass shifts specified in the list will be included in the search, so be sure to include 0 if unmodified peptides (or peptides modified only by variable modifications) are expected. Identification of mass-shifted peptides is not dependent on identification of a corresponding un-shifted peptide.
7) On the ‘PTMs’ tab, remove ‘Failed_Carbamidomethylation:-57.021464’ from the Custom mass shifts box (so that it is empty).
Set the output location and run
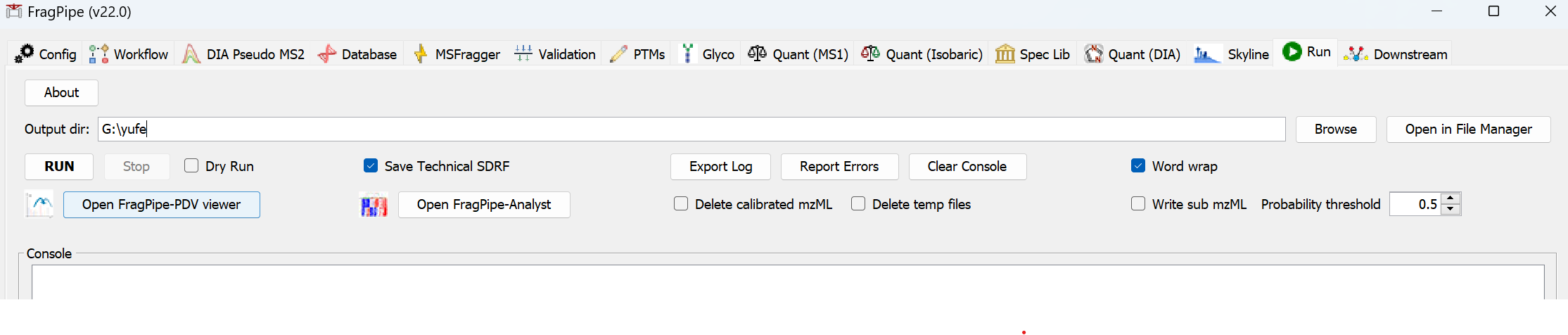
8) On the Run tab, make a new folder for the output files, then click ‘RUN’ and wait for the analysis to finish.
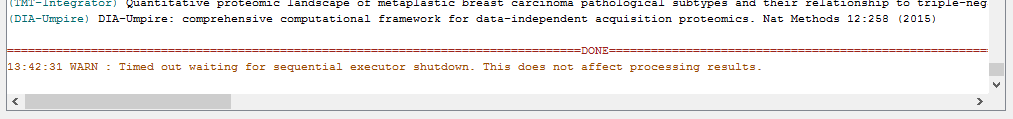
When the run is finished, ‘DONE’ will be printed at the end of the text in the console.
Examine the results
In the output location, you will see subfolders containing individual results for DMSO and PTL, plus PTM-Shepherd output files that summarize the mass shifts from both experimental conditions together. A guide to all the output files can be found here.