Diagnostic Ion Mining with PTM-Shepherd in FragPipe
Diagnostic ion mining allows allows you to find fragmentation for PTMs from offset or open searches. These can be used for things like chemical probe design or as input into MS-Fragger’s labile mode in subsequent searches. For info about the labile mode, check out the MSFragger Labile paper. For info about the diagnostic ion mining mode, check out the diagnostic ion mining paper.
This tutorial will provide a brief overview of running the diagnostic ion mining tool in FragPipe. To build a labile search workflow, we recommend loading the Diagnostic-ion-mining workflow in FragPipe. This workflow performs an open search before diagnostic ion mining, so it can be useful if you don’t know the mass of your PTM of interest. However, you can generally get more PSMs by doing an offset search if you already know the mass of your PTM, so we’ll demonstrate how to enable diagnostic ion for offset search too.
If you are new to MSFragger or FragPipe searches, please first consult the Setup and Basic tutorials.
Tutorial contents
- Diagnostic ion mining overview
- Running a diagnostic ion mining search using the default workflow
- Interpreting results
- Using PTM fragmentation patterns in subsequent labile searches
Diagnostic ion mining overview
The diagnostic ion mining module is embedded within PTM-Shepherd. It allows you to identify fragmentation patterns for PTMs identified via open searches or mass offset searches. Three types of fragmentation patterns are looked for. Each of these corresponds to an ion type that can be used in MSFragger Labile mode.
Diagnostic ions are ions that are found alone in the spectrum and have completely dissociated from the peptide. Examples of these are immonium ions derived from modified residues or oxonium ions from glycopeptides.
Peptide remainder masses are found when a peptide remains intact after the PTM fragments. It corresponds to the mass of the PTM that remains on the peptide. An example of this would be Y-ions from glycopeptides.
Fragment remainder masses are the PTM masses that remain on fragment ions (e.g., b- and y- backbone ions). If modifications are intact in the MS2, such as for non-labile modifications, then this is the mass of the modification. Sometimes, PTMs might dissociate from the peptide while taking a piece of the peptide with them, with the most famous example being the 98 Da neutral loss from Phosphoserine and Phosphothreonine. Fragment remainder masses in that case are presented as a negative remainder mass.
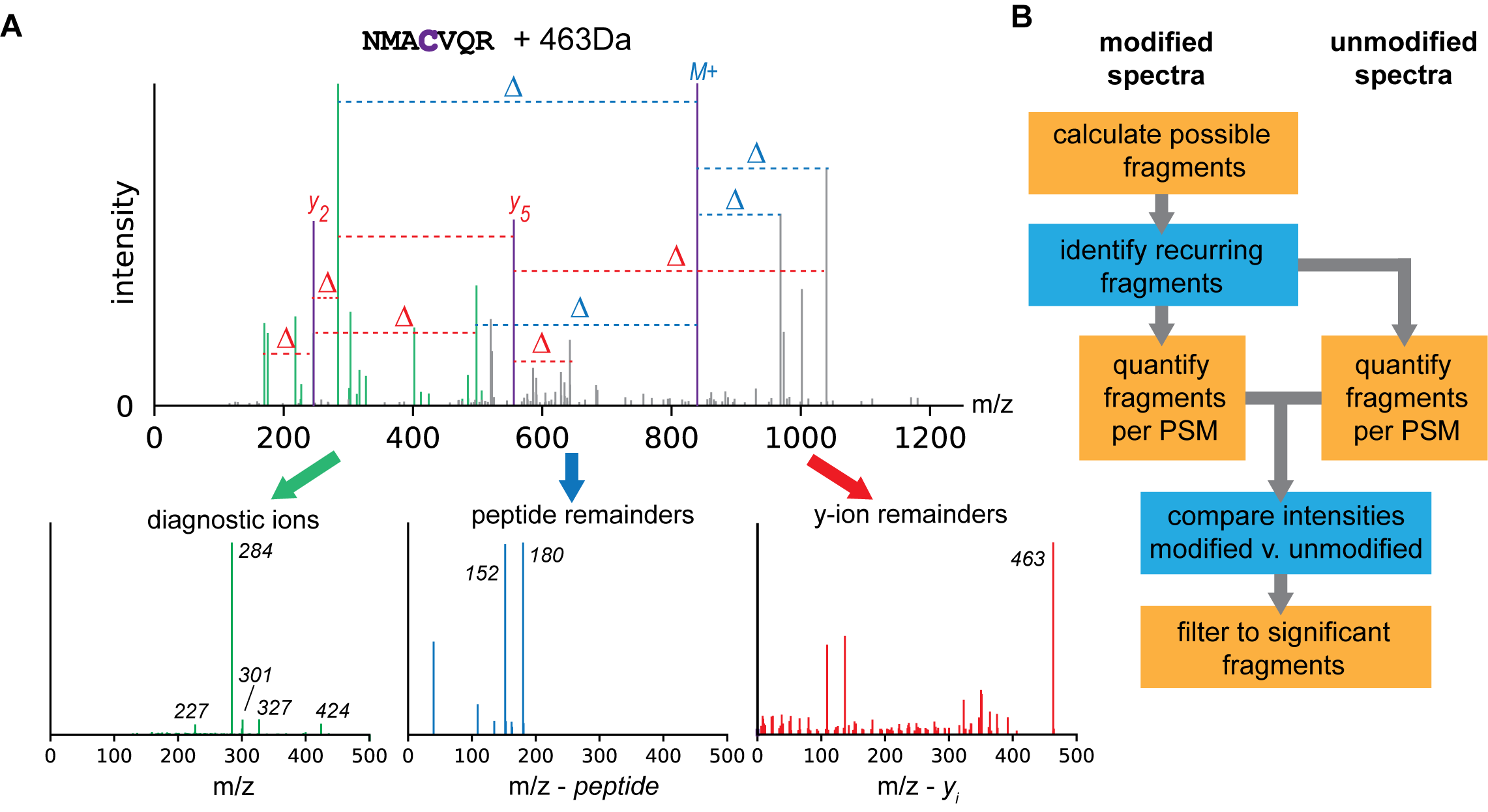
The general steps of how these are identified can be described as follows:
- Calculate every possible diagnostic ion/peptide remainder mass/fragment remainder mass for a peptide given a spectrum where it is identified
- Identify patterns that recur across peptides that bear a PTM
- Check whether these patterns are enriched for peptides bearing the PTM as compared to unmodified peptides from the same dataset
The result of this analysis is a set of diagnostic ions, peptide remainder masses, and fragment remainder masses for every mass shift in a dataset.
Running a diagnostic ion mining search using the default workflow
If you want to follow along with the tutorial and process the data yourself, you can download the input data HERE. This dataset corresponds to a photoactivatable RNA crosslinker from Photoactivatable ribonucleosides mark base-specific RNA-binding sites](https://www.nature.com/articles/s41467-021-26317-5) by Bae et al. (2021). We’re going to use the mRBS_dTamicon30K_4SU_HCD_half_1.raw file for this tutorial. Note that all of the tools we’ll be using in this analysis can directly process Thermo .raw files, so there’s no need to convert to mzML.
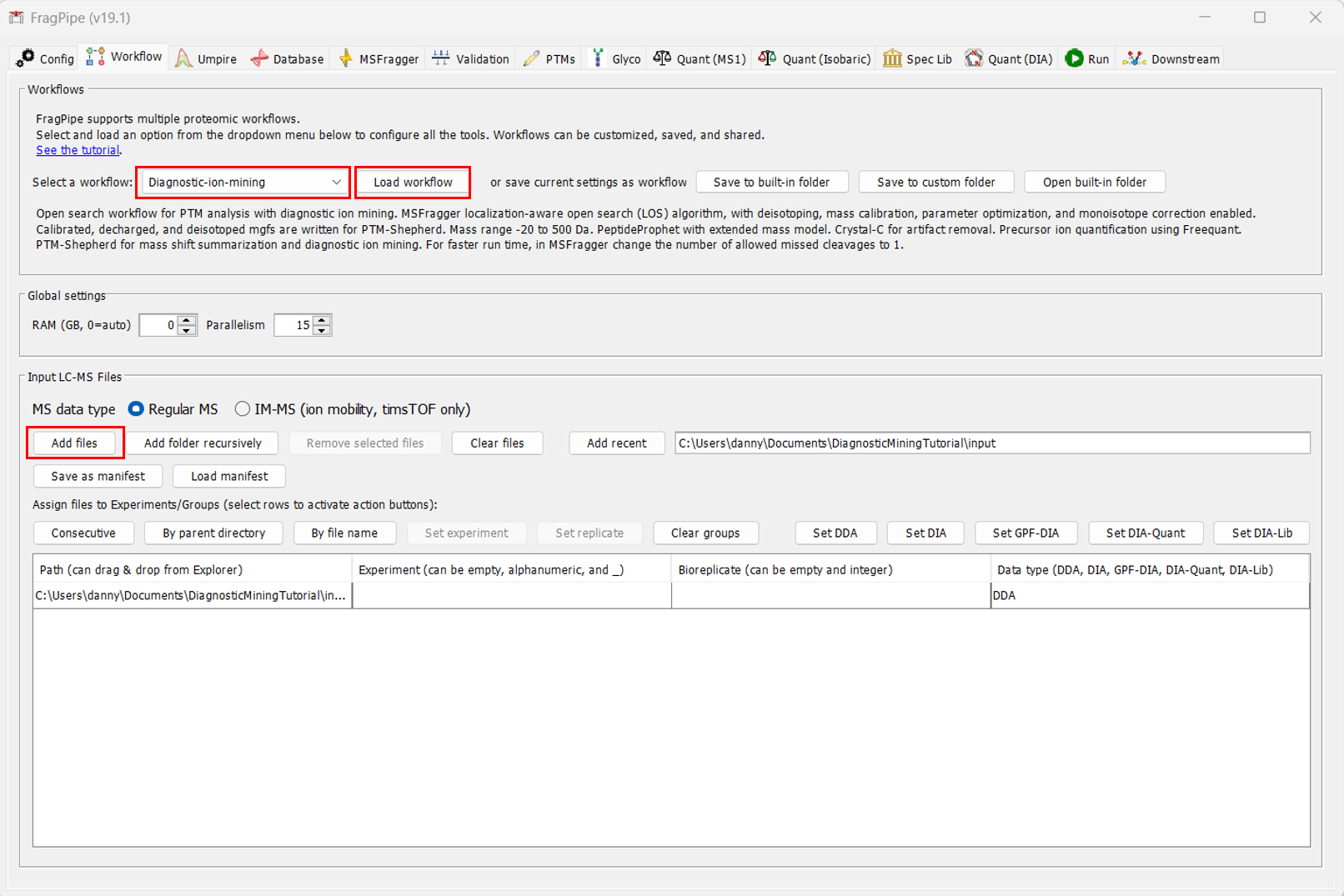
After setting up FragPipe, navigate to the Workflow tab. Upload the file to be analyzed by clicking “Add files”. Then, from the dropdown manu at the top, select the “Diagnostic-ion-mining” workflow. IMPORTANT: You must click “Load workflow” after selecting the workflow, or the parameters throughout FragPipe will not change.
Once you’ve uploaded your file and configured FragPipe for diagnostic ion mining, navigate to the “Database” tab to select your Fasta. If you don’t already have a Fasta you’d like to use, you can download one directly from Uniprot inside Fragpipe. To do that, click “Download”.
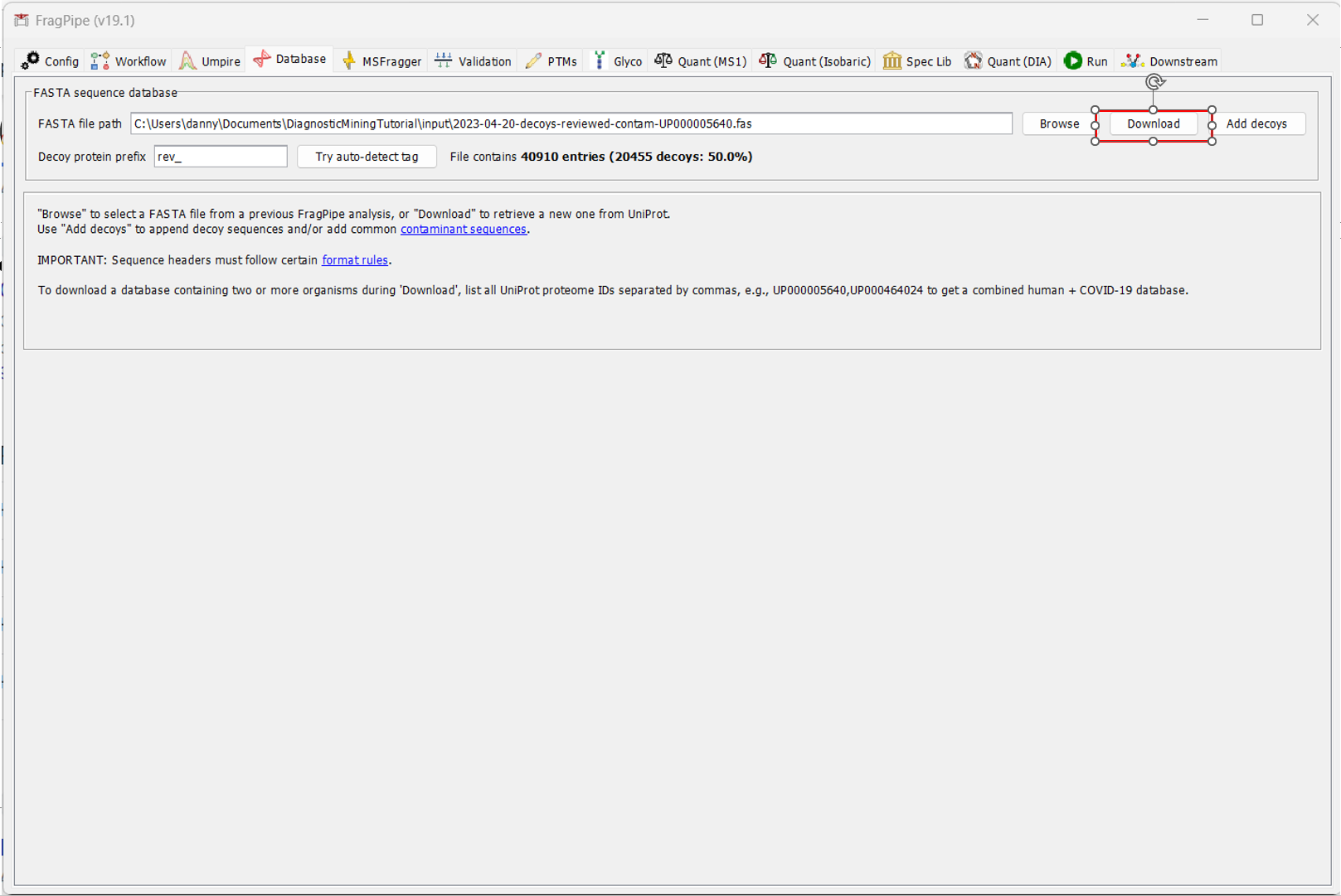
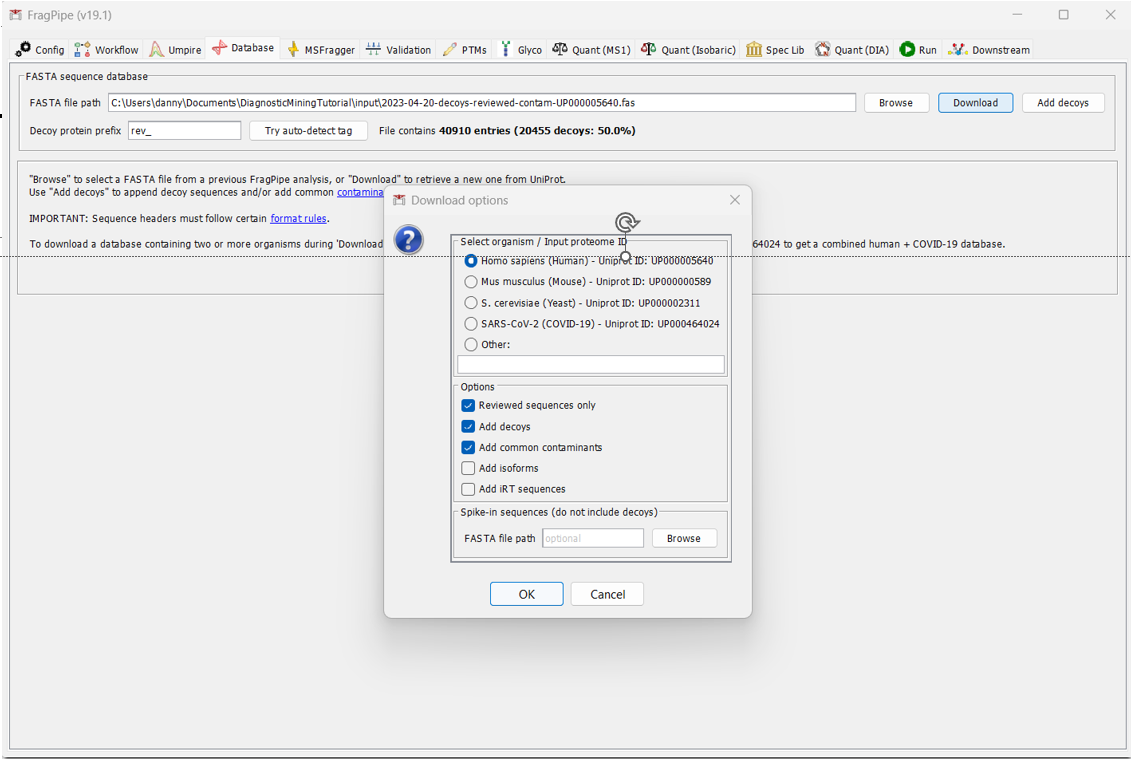
By default, FragPipe will download the reviewed Human database, add contamininant proteins to it, and generate decoys. If that all looks okay (for our analysis, it does), click OK and select the directory you would like to download it to.
That’s pretty much it! All that’s left to do is to navigate to the “Run” tab, select “Browse” to selection your ourput directory, and then click “RUN”.
Interpreting results
PTM-Shepherd created a folder called “ptm-shepherd-output” inside your output folder. The most relevant files for this analysis are the “global.profile.tsv” and the “global.diagmine.tsv”. For a description of the output tables, check out our Guide to FragPipe Results.
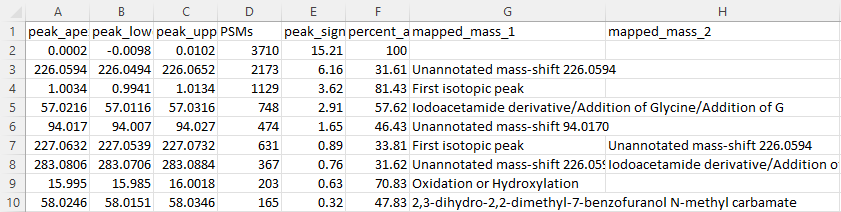
The first file we will look at is the “global.profile.tsv”. It provides several metrics based on the MS1 mass shifts detected in the dataset. The image below has both its columns and rows truncated to fit in the tutorial, but the important information for this analysis is shown.
Notably, we see two abundant mass shifts that aren’t annotated: a 226 Da mass shift and a 94 Da mass shift. These are likely to related to the analysis. Are they the same modification? To see that, we go to the next table.
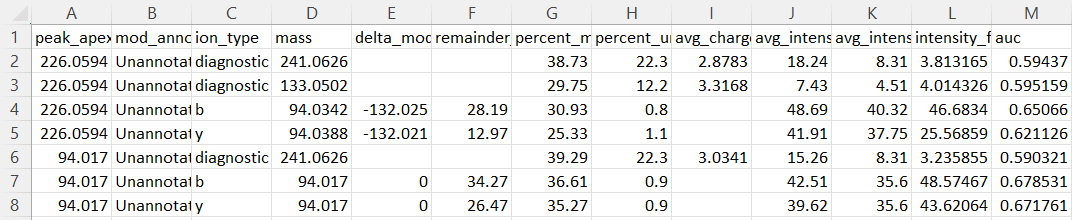
In the “global.diagmine.tsv” table, several metrics are reported to describe diagnostic fragmentation features. The two mass shifts dscribed above show similar remainder masses on b- and y-ions. This mass corresponds to the mass of the 94 Da mass shift from the MS1. Because the remainder masses produced are the same, the most reasonable explanation is that the 226 Da and 94 Da MS1 mass shifts correspond to the same modification, but there is a (226 Da - 94 Da =) 132 Da neutral loss occuring at both the MS1 and MS2 levels. The optimal way to search for these is probably an offset search with 226 Da and 94 Da as MS1 offsets, but for both only looking for the 94 Da mass on the fragment ions.
It’s also common to see diagnostic ions corresponding to PTMs. One of the diagnostic ions identified for the heavier mass shift (133 Da) corresponds to the charged version of the neutral loss described above.
Using PTM fragmentation patterns in subsequent labile searches
Now that we have some PTM fragmentaion information, we can use these patterns in the MSFragger Labile mode.