Perform two-pass search to analyze novel peptides and PTMs
FragPipe can be downloaded here. Follow the instructions on that same Releases page to launch the program. See here for help configuring FragPipe.
FragPipe has the ability to perform the “two-pass search” to detect novel peptides and PTMs semi-automatically. It performs the following three steps.
- Perform the first search to identify the canonical peptides and/or common PTMs
- Generate new “sub mzML” files by excluding the scans having good matches
- Perform the second search to identify the novel peptides and/or PTMs
Common issues of two-pass search approaches due to the limited high-quality scans in the second search
- The spectra are not mass calibrated or the mass calibration does not work well
- The rescoring tools, such as Percolator, cannot learn a good model in the second search
To address these issues, FragPipe
- performs mass calibration in the first search, and then generates sub mzML files using the calibrated spectra
- writes the Percolator models (
*_percolator.weightsfiles) to the result directory after the first search. These models will be used in the second search.
An alternaive approach to analyze novel/variant peptides
FragPipe can also perform group FDR estimation for the peptides with different levels of evidences. Please check Perform group FDR estimation for different identification groups for details.
The two-pass search approach supports the Default, LFQ-MBR, and TMT-related workflows.
Perform two-pass search for the label-free quantification data
- Load the
LFQ-MBRworkflow in theWorkflowtab - Load the LC-MS files, assign experiments and bioreplicates (if applicable), and set the
Data typetoDDA - Load your FASTA file in the
Databasetab - Go to the
Runtab, specify the output directory - [Critical] In the
Runtab, enable theWrite sub mzML. FragPipe will generate “sub mzML” files containing the scans do not pass the FDR filtering and have the probability smaller than or equal to theMin probability threshold of excluding scans from sub mzML files.
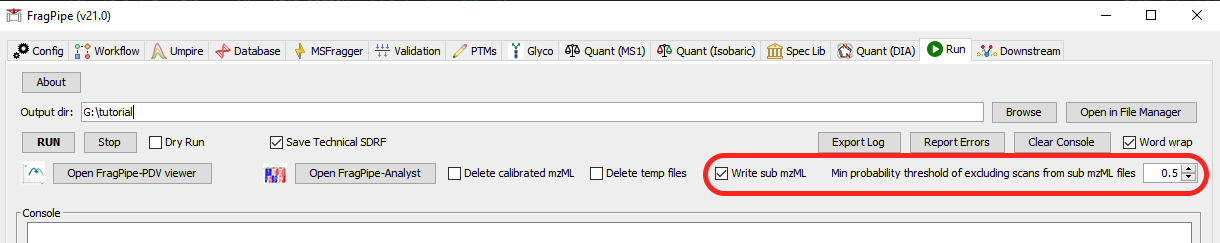
- Click run. FragPipe will perform the “first-pass search”.
- After FragPipe finish, go back to the
Workflowtab - Select the
Customworkflow and clickLoad workflow
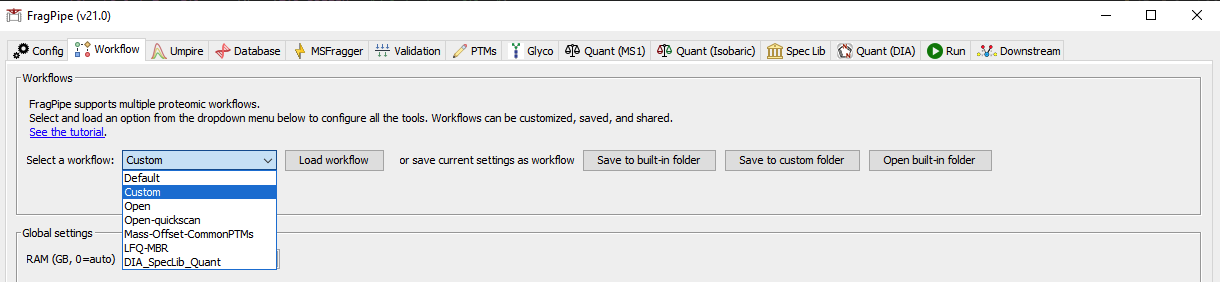
- Locate the
fragpipe-second-pass.workflowfile in the result directory and load it - Click
Clear filesto remove all LC-MS files used in the first-pass search - Click
Load manifest, locate thefragpipe-second-pass.manifestfile in the result directory, and load it
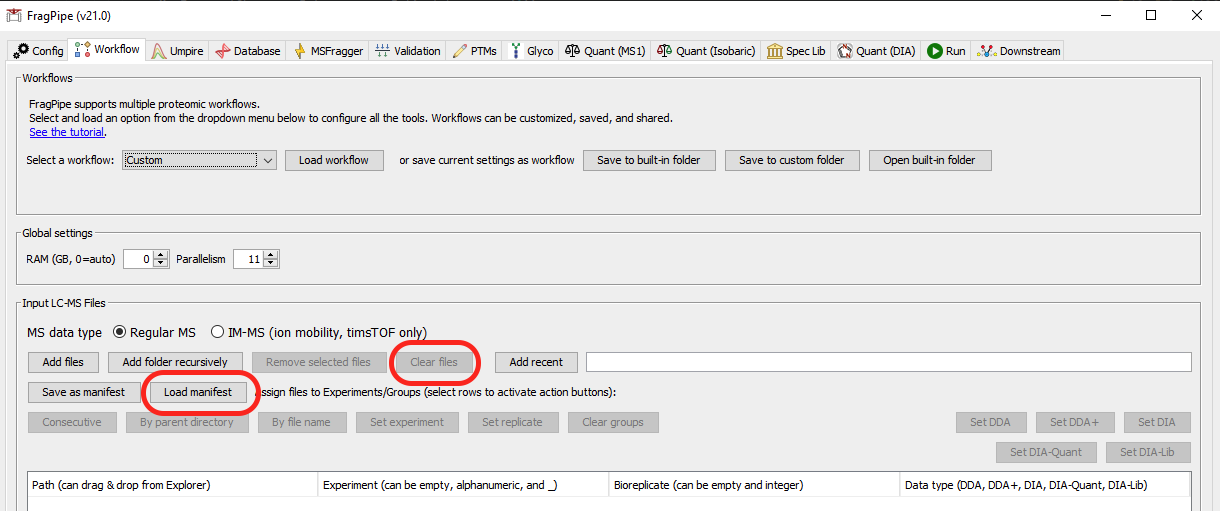
- Go to the
Databasetab to specify the FASTA file for the second search. - Go to
Runtab, specify the new output directory and clickRun
Perform two-pass search for the isobaric-labeling data
- Load one of the TMT workflow in the
Workflowtab - Load the LC-MS files, assign experiments and bioreplicates (if applicable), and set the
Data typetoDDA - Load your FASTA file in the
Databasetab - Go to the
Quant (Isobaric)tab, configure the sample/channel annotations. Details can be found in the other tutorial - Go to the
Runtab, specify the output directory - [Critical] In the
Runtab, enable theWrite sub mzML. FragPipe will generate “sub mzML” files containing the scans do not pass the FDR filtering and have the probability smaller than or equal to theMin probability threshold of excluding scans from sub mzML files.
- Click run. FragPipe will perform the “first-pass search”.
- After FragPipe finish, go back to the
Workflowtab - Select the
Customworkflow and clickLoad workflow
- Locate the
fragpipe-second-pass.workflowfile in the result directory and load it - Click
Clear filesto remove all LC-MS files used in the first-pass search - Click
Load manifest, locate thefragpipe-second-pass.manifestfile in the result directory, and load it
- Go to the
Databasetab to specify the FASTA file for the second search. - Go to the
Quant (Isobaric)tab to ensure that the sample/channel annotation is correct. - Go to
Runtab, specify the new output directory and clickRun
Key References
Desai H, Ofori S, Boatner L, Yu F, Villanueva M, Ung N, Nesvizhskii AI, Backus K. Multi-omic stratification of the missense variant cysteinome, bioRxiv (2023).