Analyzing DIA data
FragPipe can be downloaded here. Follow the instructions on that same Releases page to launch the program. See here for help configuring FragPipe.
FragPipe currently offers two workflows for DIA data:
1) DIA_SpecLib_Quant - takes DIA data (plus optional DDA data) as input, builds a spectral library using MSFragger-DIA, then quantifies with DIA-NN
2) DIA_DIA-Umpire_SpecLib_Quant - takes DIA data (plus optional DDA data) as input, DIA-Umpire generates pseudo-MS/MS spectra from the DIA files (instead of direct search with MSFragger-DIA), then MSFragger in DDA mode is used to search, followed by quantification with DIA-NN
Please note:
- DIA data acquired with overlapping/staggered windows must be converted to mzML using the ‘Demultiplex’ filter, see this page.
- To quantify from .raw files, Thermo MS File Reader must be installed, see the DIA-NN documentation for details.
- Any pseudo-MS/MS files from DIA-Umpire (
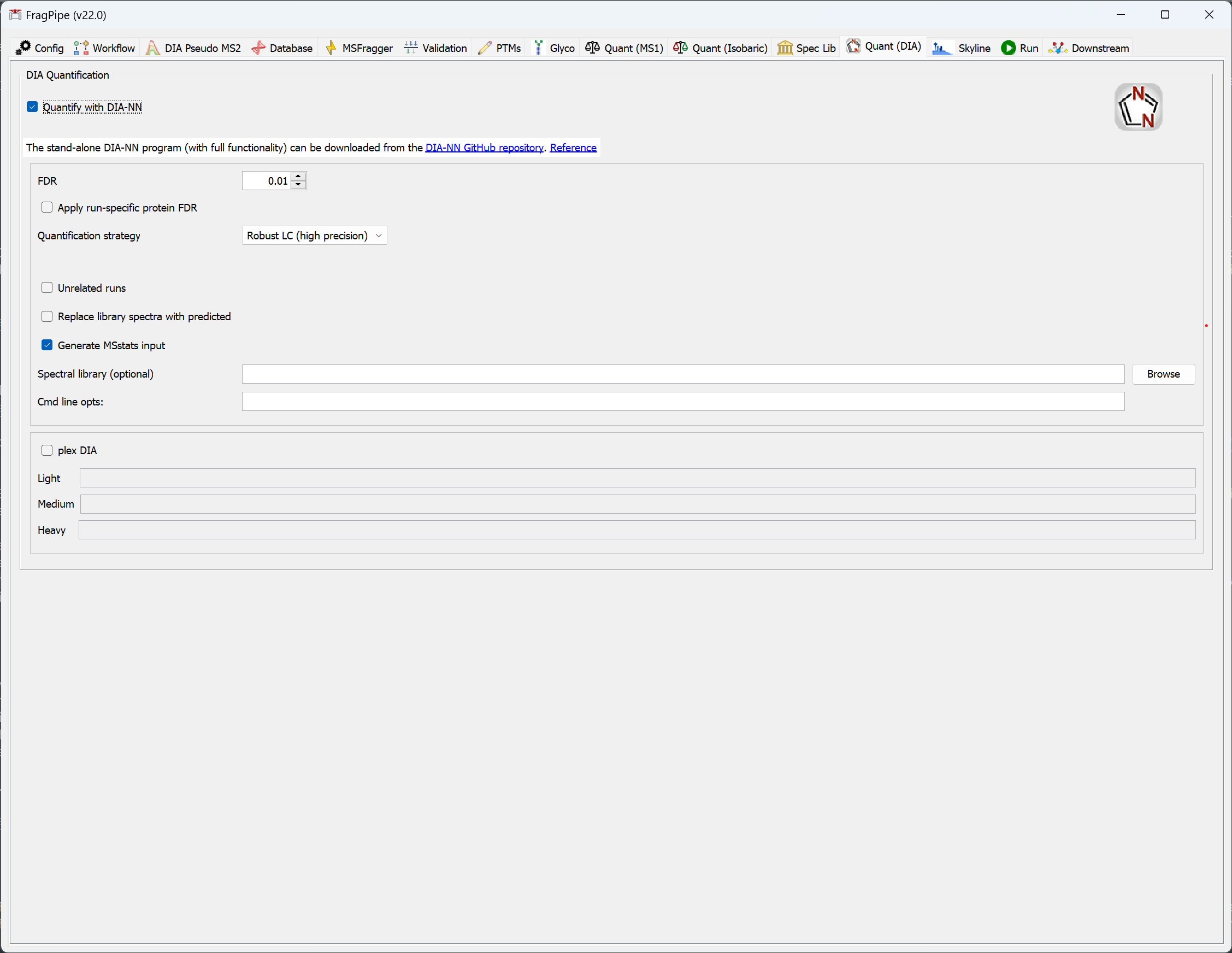
*_Q1.mzML,*_Q2.mzML,*_Q3.mzML) should be designated DDA data type on the Workflow tab. - If you already have a spectral library and want to quantify only, uncheck all steps except ‘Quantify with DIA-NN’ on the ‘Quant (DIA)’ tab, set the path to the spectral library, and run.
- If iRT peptides were spiked-in to the samples, change the ‘RT calibration’ option on the ‘Spec Lib’ tab to ‘iRT’. EasyPQP will use the ciRT option by default.
- Skyline users may also choose to import interact-.pep.xml files into Skyline for spectral library building and further analysis of DIA experiments, see this tutorial.
In this tutorial, We will analyse a subset of the dataset published in Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. In the original studies, researchers from the CPTAC (Clinical Proteomic Tumor Analysis Consortium) profiled tumor (T) samples, together with normal adjacent tissue (NAT) samples from each cancer patient, to understand the tumorigenesis of ccRCC. 110 tumor and 83 NAT samples were collected from patients and their proteomes were profiled via mass spectrometry. These samples were originally profiled using: i) tandem mass tag (TMT), and ii) data-independent acquisition (DIA). The DIA set was generated on an Orbitrap Lumos mass spectrometer with a variable window acquisition scheme.
Here, we will use just 10 DIA runs from 5 ccRCC patients, one tumor and one paired NAT sample for each patient. To make the data processing faster, we will use only data in two isolation windows (613 to 650 Th mass range) from each original mzML file. The files can be downloaded from here.
Configure FragPipe
Python (with EasyPQP installed) is needed for spectral library generation. On the Config tab, check that a valid Python path is provided (Python version will be shown) and that EasyPQP is ‘Available’. If Python is installed but EasyPQP is missing, click the ‘Install/Upgrade EasyPQP’ button and wait a minute or so for installation. For help installing Python, see this page.
DIA_SpecLib_Quant
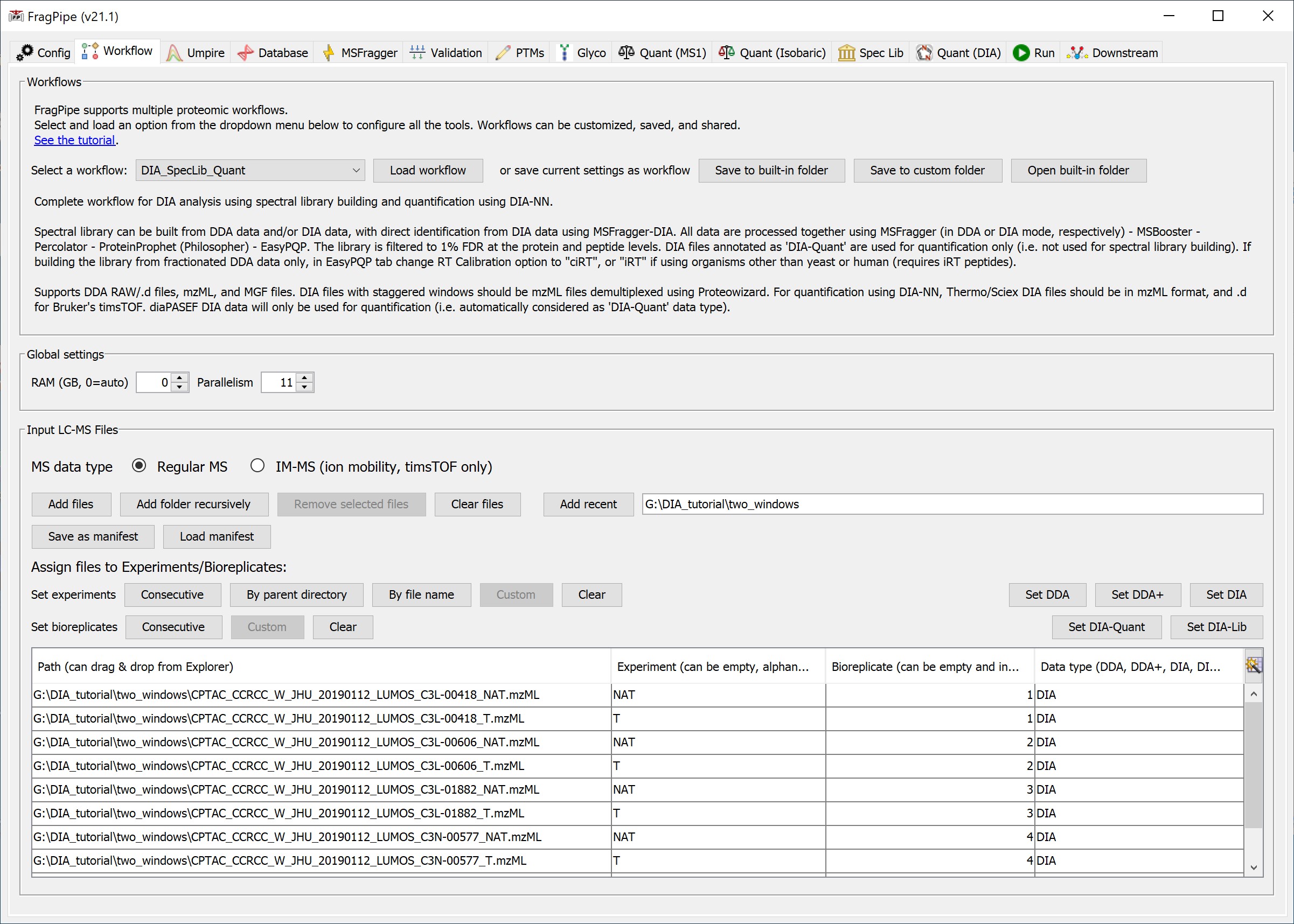
- In the Workflow tab, select the ‘DIA_SpecLib_Quant’ workflow from the dropdown menu and click ‘Load’.
- Load DIA (and optionally additional DDA) spectral files in mzML or raw format. The data type of each file will be automatically guessed by FragPipe. The guess is based on the folder and file name. Please double check the data type and adjust it if necessary.
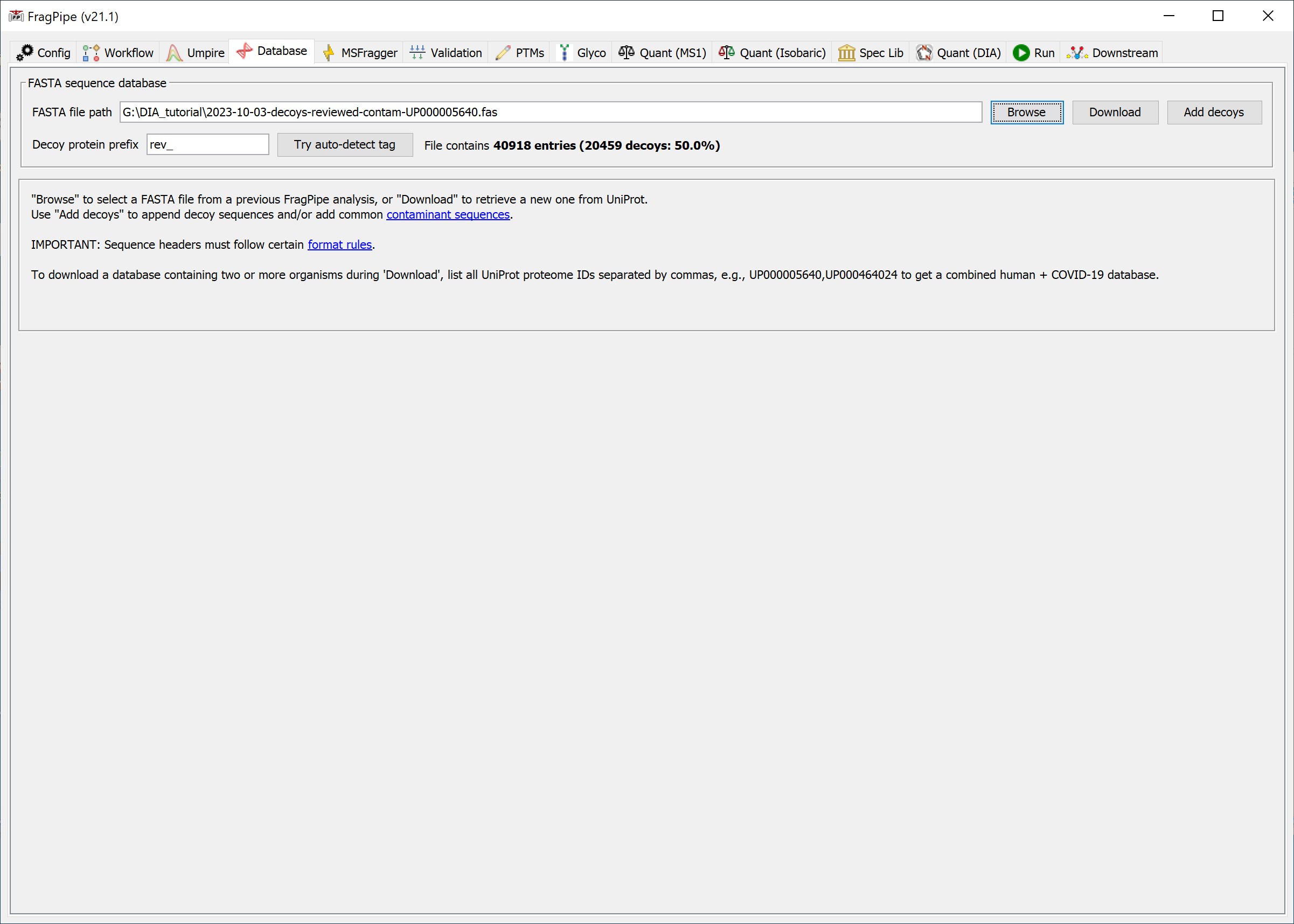
- On the Database tab, use ‘Browse’ to select the FASTA sequence database file.
- On the ‘Quant (DIA)’ tab, note that DIA-NN will be run unless unchecked. The spectral library generated by FragPipe will automatically be passed to DIA-NN for quantification of the DIA files provided.
- On the ‘Run’ tab, set the output directory and click ‘Run’.
DIA_DIA-Umpire_SpecLib_Quant
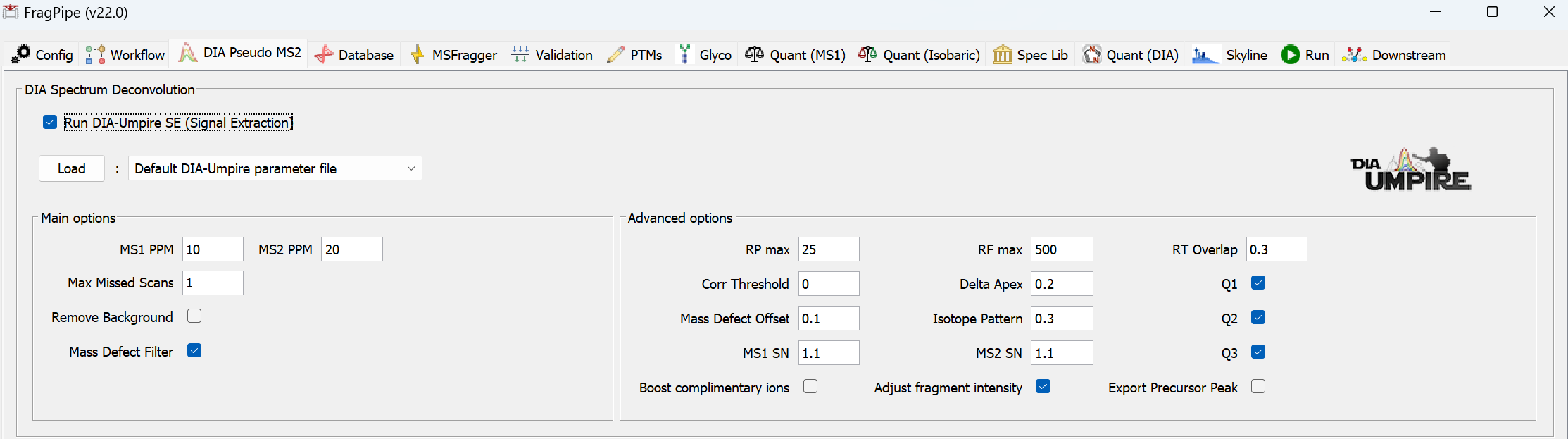
Follow the same steps as above to run the ‘DIA_DIA-Umpire_SpecLib_Quant’ workflow, which uses DIA-Umpire to generate pseudo-DDA spectra instead of MSFragger-DIA direct search of the DIA files. On the ‘Umpire’ tab, choose the appropriate settings:
- Change ‘Max Missed Scans’ to 2 if building a library from DIA data only (slower run time but higher identification sensitivity).
- Check ‘Remove Background’ if building a hybrid DDA+DIA library (see below) and if there are many DIA runs (fastest run time).
- Uncheck ‘Mass Defect Filter’ if DIA data is generated on modification-enriched peptides (e.g. phospho), or if you’re interested in extended PTM searches.
Please note that if DIA-Umpire fails or is interrupted, temporary files will cause issues if the process runs again. Make sure to delete any temporary files that are generated alongside the raw/mzML files before re-running FragPipe.
A more comprehensive tutorial from the EMBO Practical Course: Targeted proteomics
The tutorial file can be found from here.
Key References
Yu F, Teo GC, Kong AT, Fröhlich K, Li GX, Demichev V, Nesvizhskii AI. Analysis of DIA proteomics data using MSFragger-DIA and FragPipe computational platform, Nature Communications 14:4154 (2023).
Tsou CC, Avtonomov D, Larsen B, Tucholska M, Choi H, Gingras AC, Nesvizhskii AI. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics, Nature Methods 12:258-64 (2015).
Demichev V, Szyrwiel L, Yu F, Teo GC, Rosenberger G, Niewienda A, Ludwig D, Decker J, Kaspar-Schoenefeld S, Lilley KS, Mülleder M, Nesvizhskii AI, Ralser M. dia-PASEF data analysis using FragPipe and DIA-NN for deep proteomics of low sample amounts, Nature Communications 13:3944 (2022).